Unravelling 3D genome organisation
The first paper of my PhD is finally out! Its full title is "Integrative modeling reveals the principles of multi-scale chromatin boundary formation in human nuclear organization" but in talks and posters I've been referring to the project as "Unravelling higher order chromatin organisation" (geddit?) or even "the ENCODE-ing of ..." (referencing ENCODE, of course).
This project was done entirely with open data, I've made my code open source and in the interests of open science I thought I'd write up a blog summary of the paper.
Data
The project came out of the observation (made a couple of years ago now) that Hi-C datasets were available for each of the tier 1 ENCODE human cell lines: meaning we had access to an unprecedented amount of matched chromatin data.



Though now better sources are available, when we started there were three human Hi-C papers we gathered data from : the original Hi-C paper (Leiberman Aiden et al., 2009); an improved method, TCC, with also ran Hi-C for comparison (Kalhor et al., 2011); and a much more-deeply sequenced dataset that first reported TADs (Dixon et al., 2012). These papers produced genome-wide Hi-C data for human cell types K562, Gm12878 and H1 hESC respectively, and as mentioned, these are the ENCODE tier 1 cell lines.

These Hi-C datasets were produced by different labs, using slightly different protocols and with varying levels of sequencing. With this in mind, directly comparing contacts is going to be tricky, even after thorough normalisation. In our analysis, we relied mostly on abstracted measures of chromatin conformation: compartment eigenvectors, TAD calls or within cell-line contacts only.
Analysis
Broadly we were interested in how chromatin features are related to genome conformation. The main questions we try to address in the manuscript boil down to:
To what extent does higher order chromatin structure — by which I mostly mean megabase-sized topological domains and multi-megabase chromosome compartments — vary between cell types, and is there interesting biology where they're discordant?
How do chromatin features (like histone modifications, transcription factors) connect to what's going on at the level of higher-order chromatin structure? What can we learn the rules underlying this relationship?
How do the boundary regions demarcating higher-order domains vary between cell types?
1. Genome organisation pretty similar across human cell types
After re-processing each Hi-C data uniformly from raw sequencing reads (and applying iterative correction), it wasn't a huge suprise to find a broad agreement of higher-order chromatin structure between human cell types.1
Along the left-hand side of figure 1 I'm plotting the principal component eigenvector derived from Hi-C contact maps (reflecting A/B compartment profile, following Leiberman Aiden et al., 2009). You can see cell type profiles track one another along the chromosome, the genome-wide Pearson correlation of these values a 1 Mb resolution was around 0.80.
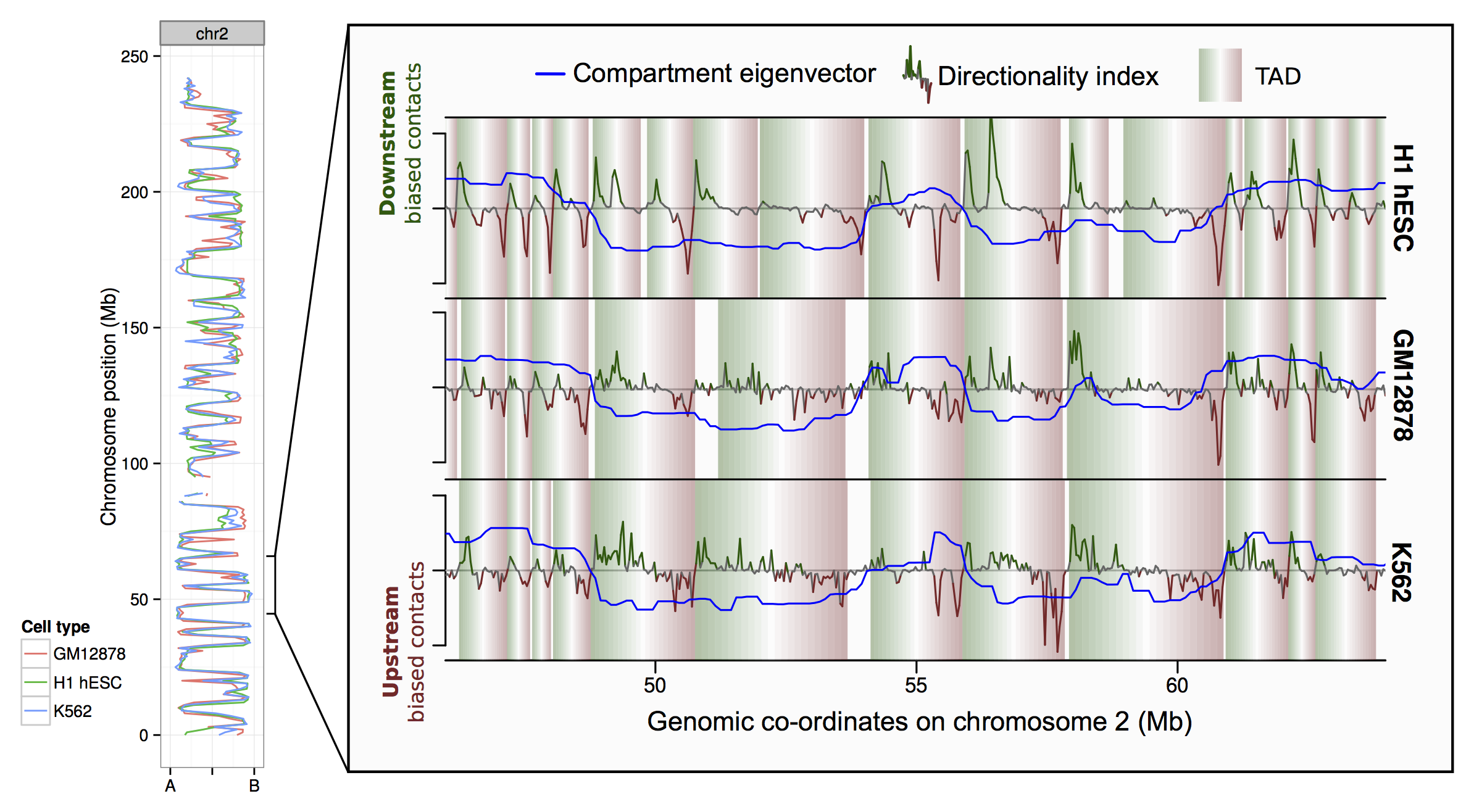
Similarly at the level of TADs — shaded rectangles in the zoomed region — there's a decent agreement between cell types. Despite the sub-optimal TAD-calling algorithm (the published Dixon et al., 2012) and a sparse contact map given the resolution (particularly in K562), there was still around a third of boundaries in one cell type that could be very precisely mapped to boundaries in the other two.
2. Chromatin features predict aspects of genome architecture
In short we built Random Forest regression models to predict the continuous compartment profile (the blue line in the above figure), and it achieved high accuracy. This wasn't wholly surprising: strong correlations between individual input variables and compartment profile has already been reported (even in the original Leiberman Aiden paper), so the interest really comes from dissecting and cross-applying these models, rather than from their raw metrics.
{kind=link}
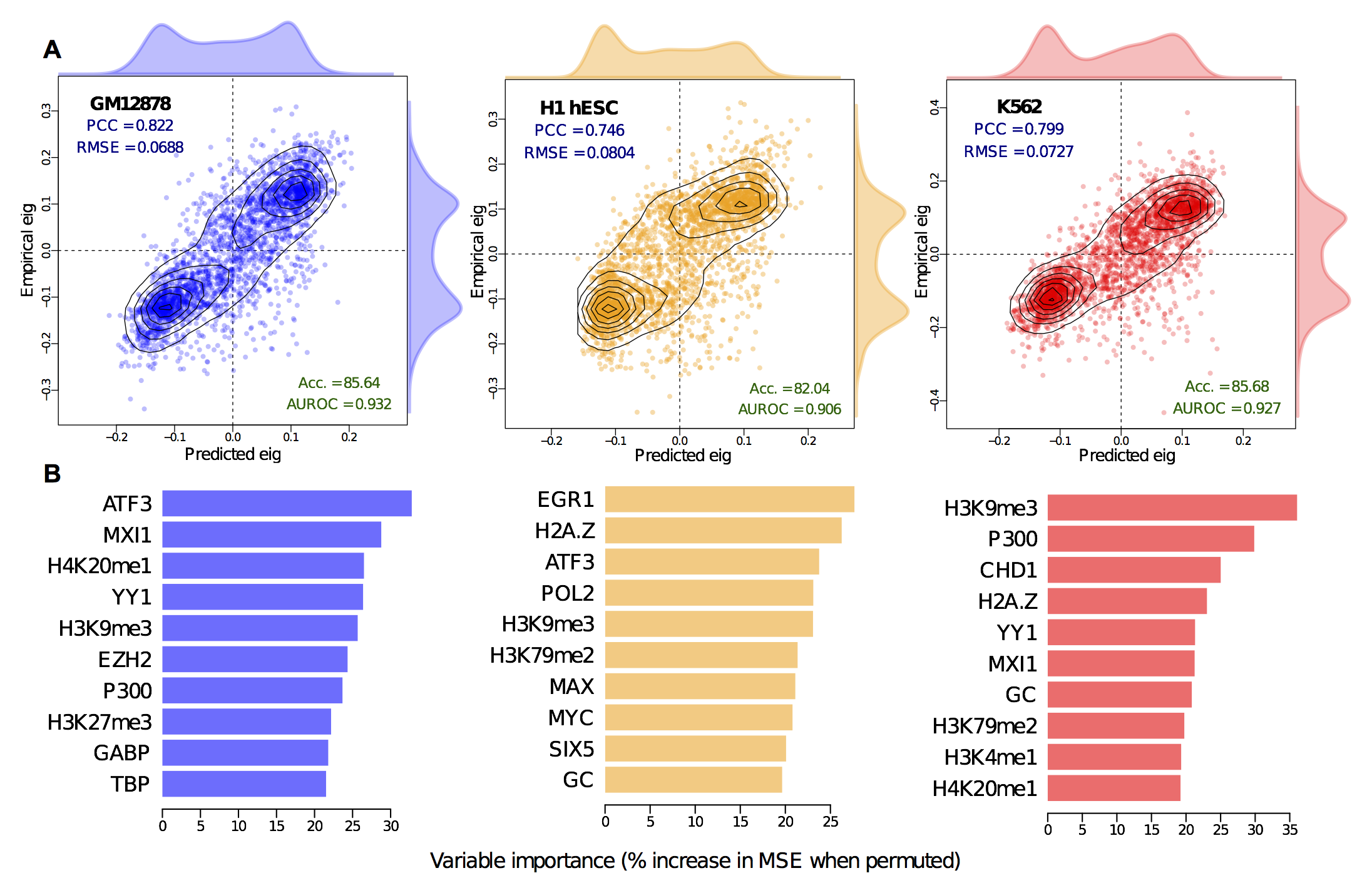
We found that models learned in one cell-type had almost as much predictive power in another cell type, suggested common rules governing higher order structure. However, we've already seen that much of this structure is the same between cell types, so are these fixed regions inflating our accuracy measures?
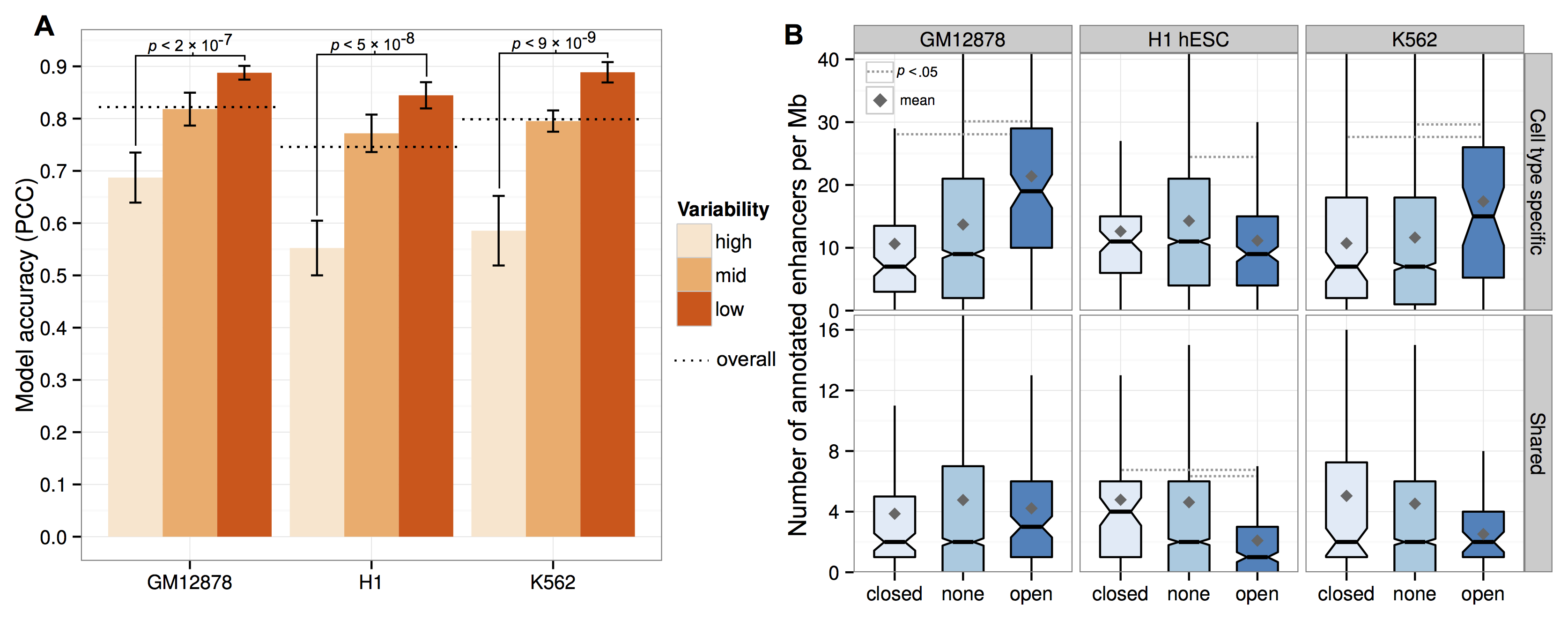
It turns out that yes, if you split your input dataset by how variable these regions are between cell types, it turns out those most variable regions are more difficult to predict (A, above).
This raises another question: are these variable regions in some way more noisy or is there something interesting going on? The right panel (B, above) tries to address this by doing some functional analysis. The results were that "flipped" open regions (e.g. a megabase active in Gm12878, but a closed, repressive compartment in the other two human cell types) showed an enrichment for predicted enhancer chromatin states,2 potentially highlighting areas of cell-type specific gene activation. We followed this up by picking out some examples (A, below) and returning to Hi-C contacts to look for changes in long-range interactions.
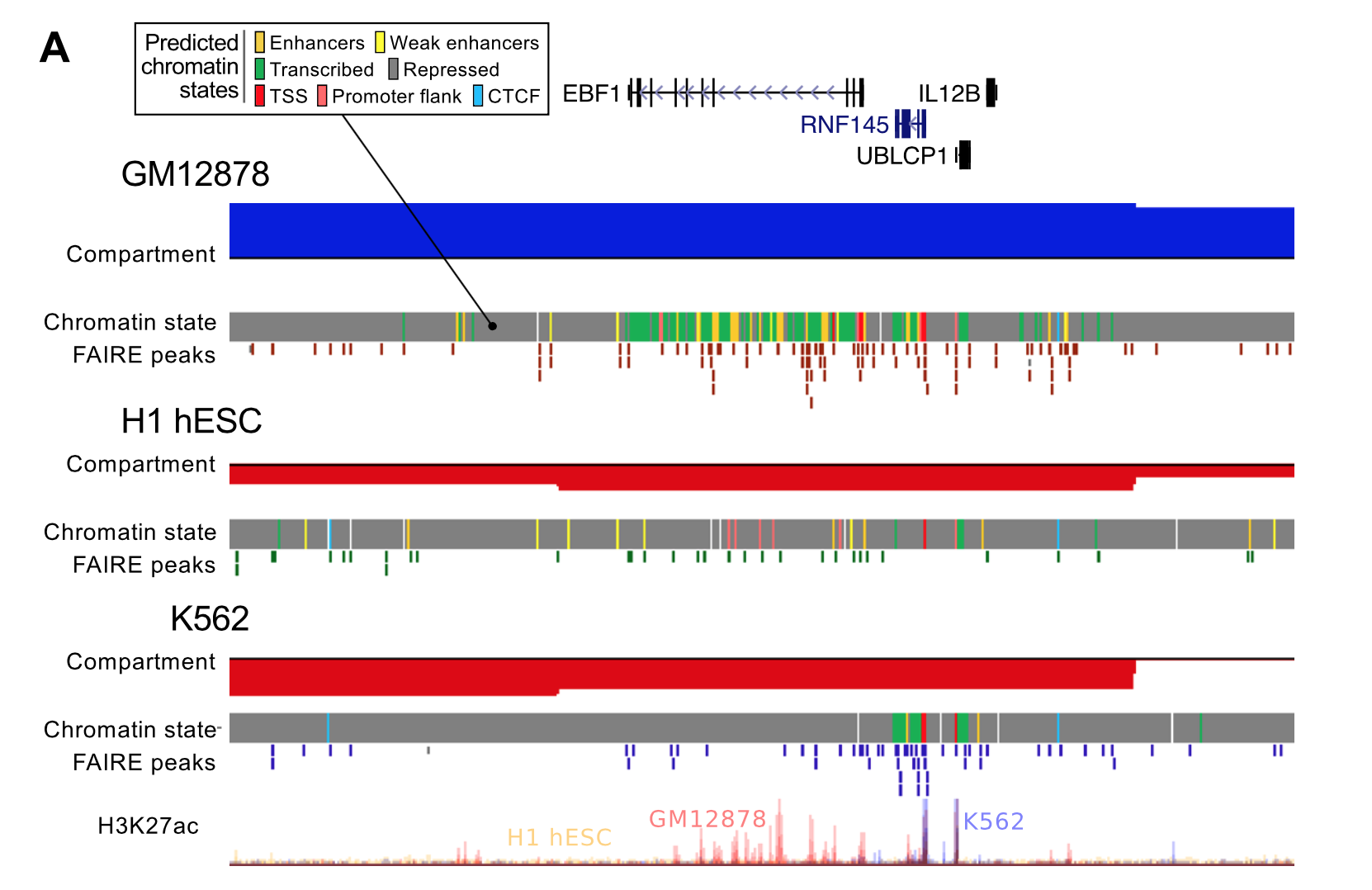
3. Domain boundaries enriched for lots of things
The last part of the paper I want to mention was an analysis of domain boundary enrichments. TAD boundaries were brought to the fore by Dixon et al. (2012), but there's been debate about how important or interesting these spacers are. Are boundaries responsible for domains (i.e. binding of insulators and other factors blocks heterochromatin expansion) or are boundaries just "what's left" when an active domain is adjacent to, say, a repressive lamina-associated domain?
While we couldn't resolve that debate in this paper, we were still interested in comparing TAD boundaries across cell types, as well as looking at compartment boundaries too. We also statistically tested the enrichment of each feature, rather than just showing the "average-o-gram" profiles.
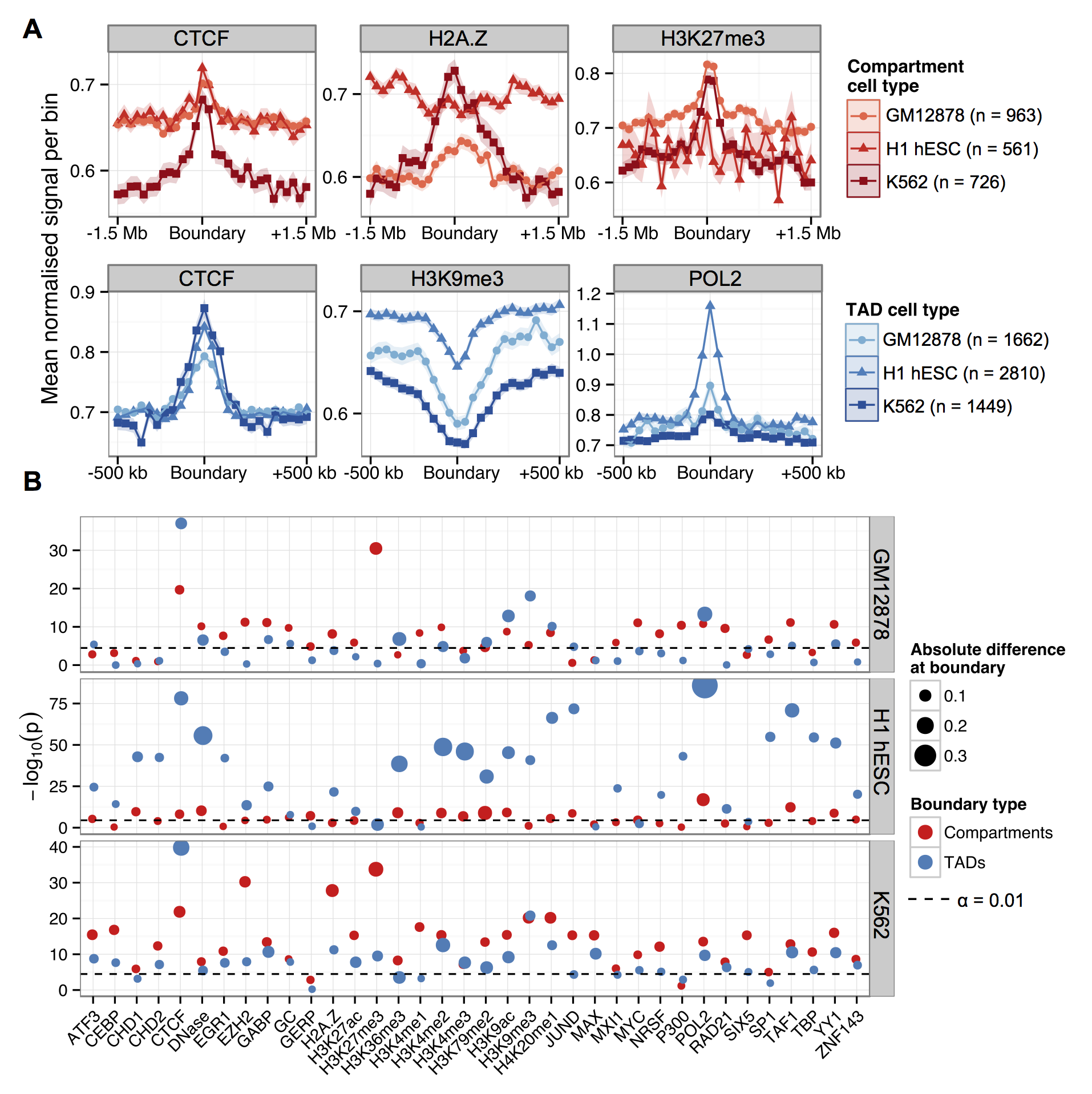
The results recap some of the known TAD enrichments (CTCF, Pol2) but also find an overlapping range of peaked features at compartment boundaries, and at a broader scale.
Summary
Overall we found higher order chromatin structure is well-conserved between human cell types and well-described by locus-level chromatin features, but divergent regions contain cell-type-specific biology that could be cause or consequence of these differences. Our difficulties predicting these more flexible regions could be due to missing input features from our datasets or, more likely in my view, local activation and repression mechanisms not captured by a genome-wide model.
Links
I've given a very brief overview of a lot of work, please do check out the paper if interested. It comes with the obligatory lengthy supplementary materials full of more good stuff.
Code to reproduce all analyses shown in the paper and supplementary is avaiable on github: github.com/blmoore/3dgenome
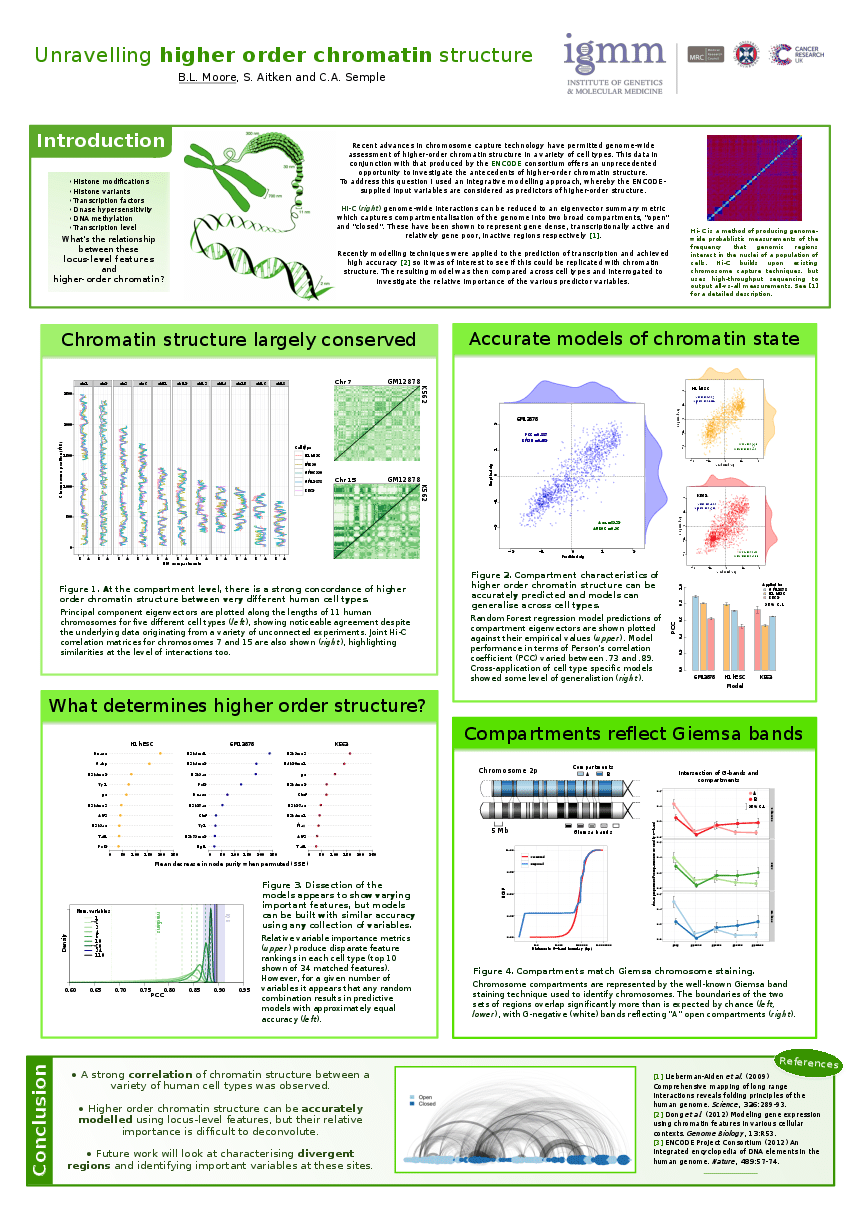
Posters related to this project have been shared on figshare.
Slides are available for a talk I gave at a local research group meet-up.
-
Of course these are probabilistic frequencies derived from cell populations rather than the individual cell comparisons achievable with single cell Hi-C ↩
-
Predicted by
ChromHMMandSegWayconsensus annotations ↩